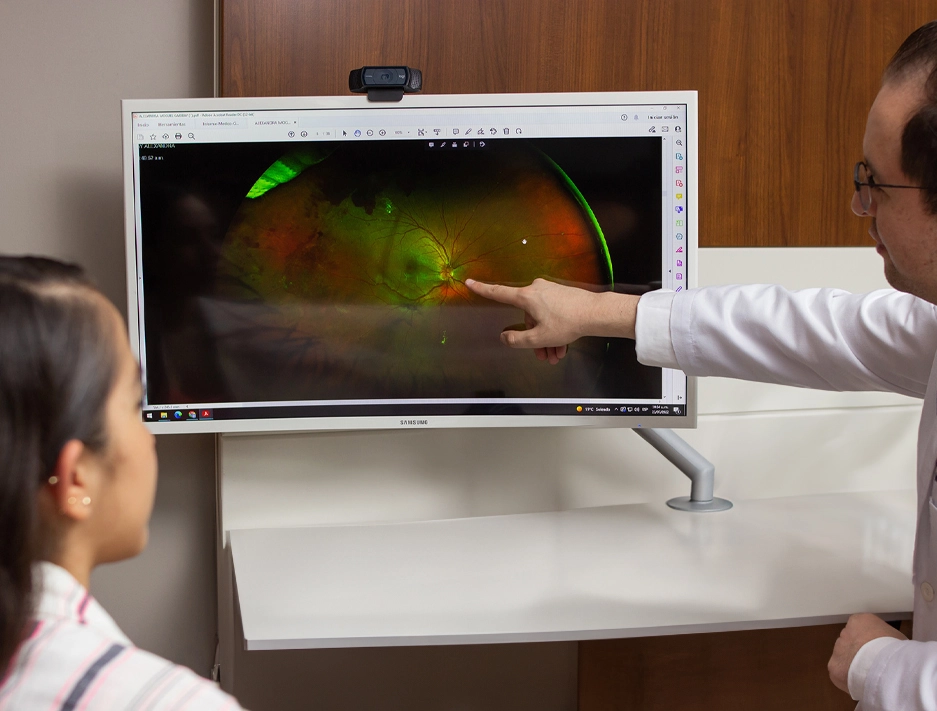
La retinosis pigmentaria es la distrofia de retina más común a nivel mundial. Se trata de una enfermedad hereditaria que destruye progresivamente los fotorreceptores de la retina —principalmente los bastones— afectando primero la visión nocturna y periférica, y avanzando con el tiempo hacia la visión central. El diagnóstico temprano y el seguimiento por un especialista en enfermedades hereditarias de la retina son determinantes para preservar la función visual el mayor tiempo posible.
La retinosis pigmentaria (RP), también conocida como retinitis pigmentaria, es una enfermedad genética y hereditaria que causa la degeneración progresiva de los fotorreceptores de la retina. Según la American Academy of Ophthalmology (AAO), afecta aproximadamente a 1 de cada 4,000 personas en el mundo, lo que la convierte en la causa más frecuente de ceguera hereditaria en adultos en edad laboral.
A diferencia de enfermedades como la retinopatía diabética —que surge por complicaciones sistémicas— o la degeneración macular asociada a la edad, la retinosis pigmentaria tiene un origen genético desde el nacimiento. Los síntomas pueden aparecer en la infancia, adolescencia o adultez temprana, y su progresión varía enormemente según la mutación genética específica de cada persona.
La retinosis pigmentaria pertenece al grupo más amplio de las distrofias de retina, que engloba más de 250 enfermedades genéticas distintas que afectan la retina.
La retina tiene dos tipos principales de fotorreceptores: los bastones y los conos. Los bastones son responsables de la visión en condiciones de poca luz y de la visión periférica. Los conos se encargan de la visión central y la percepción del color.
En la retinosis pigmentaria, los bastones son los primeros en degenerarse. Por eso los síntomas iniciales suelen ser nocturnos y periféricos. Con el tiempo, la pérdida puede avanzar hacia los conos, comprometiendo también la visión central —aunque muchos pacientes conservan algo de visión funcional durante toda su vida.
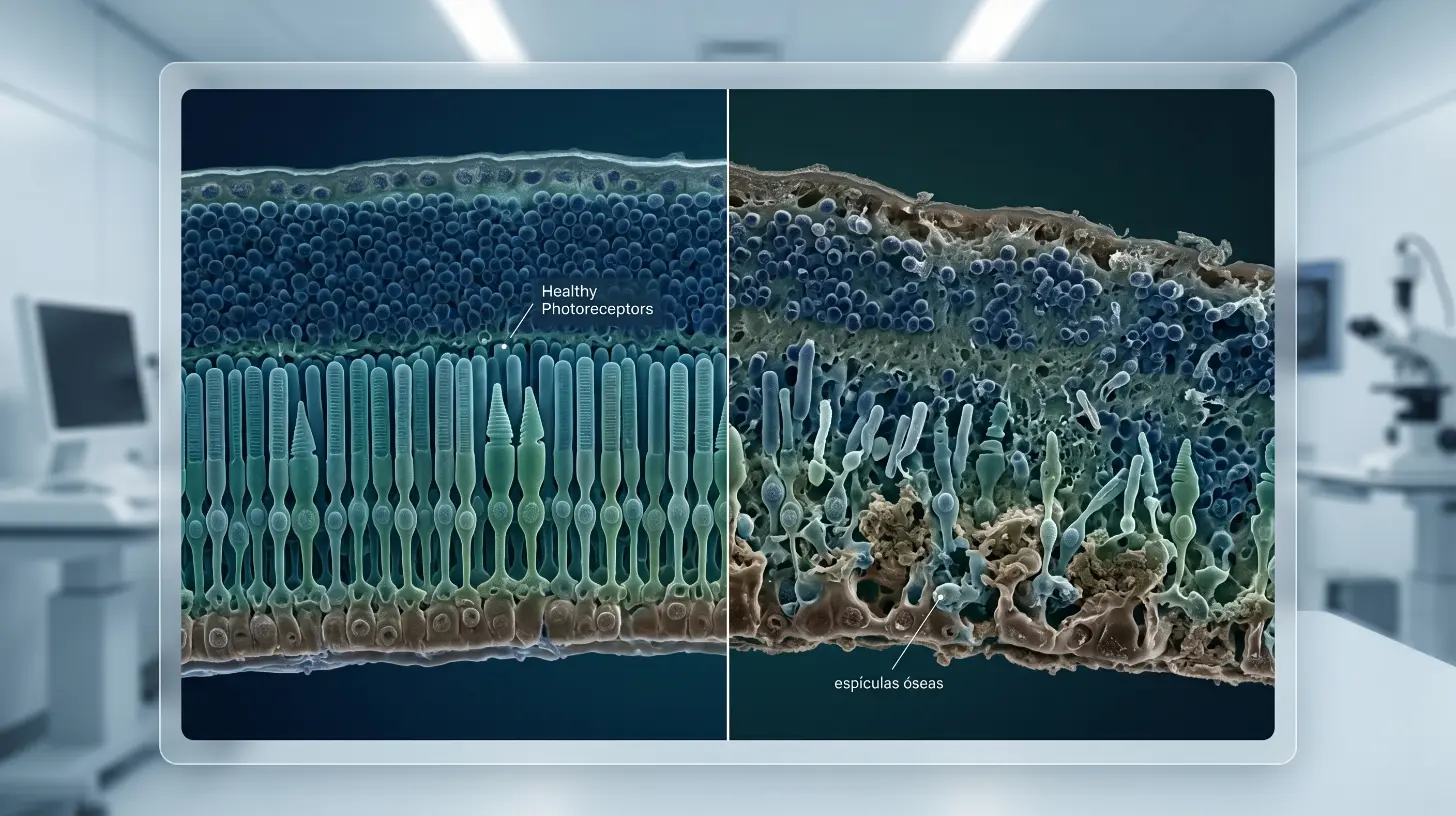
Los síntomas varían según la etapa de la enfermedad y la mutación genética específica. Según el National Eye Institute (NEI), los más frecuentes son:
Etapa temprana:
Etapa intermedia:
Etapa avanzada:
Es importante destacar que la progresión es lenta y variable. Muchos pacientes con retinosis pigmentaria conservan visión funcional durante décadas, especialmente cuando reciben seguimiento especializado.

La retinosis pigmentaria es causada por mutaciones en más de 100 genes diferentes que afectan el funcionamiento de los fotorreceptores o del epitelio pigmentario de la retina. Esta diversidad genética es precisamente la razón por la que la enfermedad tiene presentaciones tan distintas entre pacientes.
Los patrones de herencia más comunes son:
Identificar el patrón de herencia y la mutación específica es fundamental, ya que determina el riesgo para otros familiares, el pronóstico visual y la elegibilidad para tratamientos como la terapia génica.
A diferencia de distrofias maculares como la enfermedad de Stargardt, que afecta primero la visión central, la retinosis pigmentaria compromete inicialmente la visión periférica y nocturna.
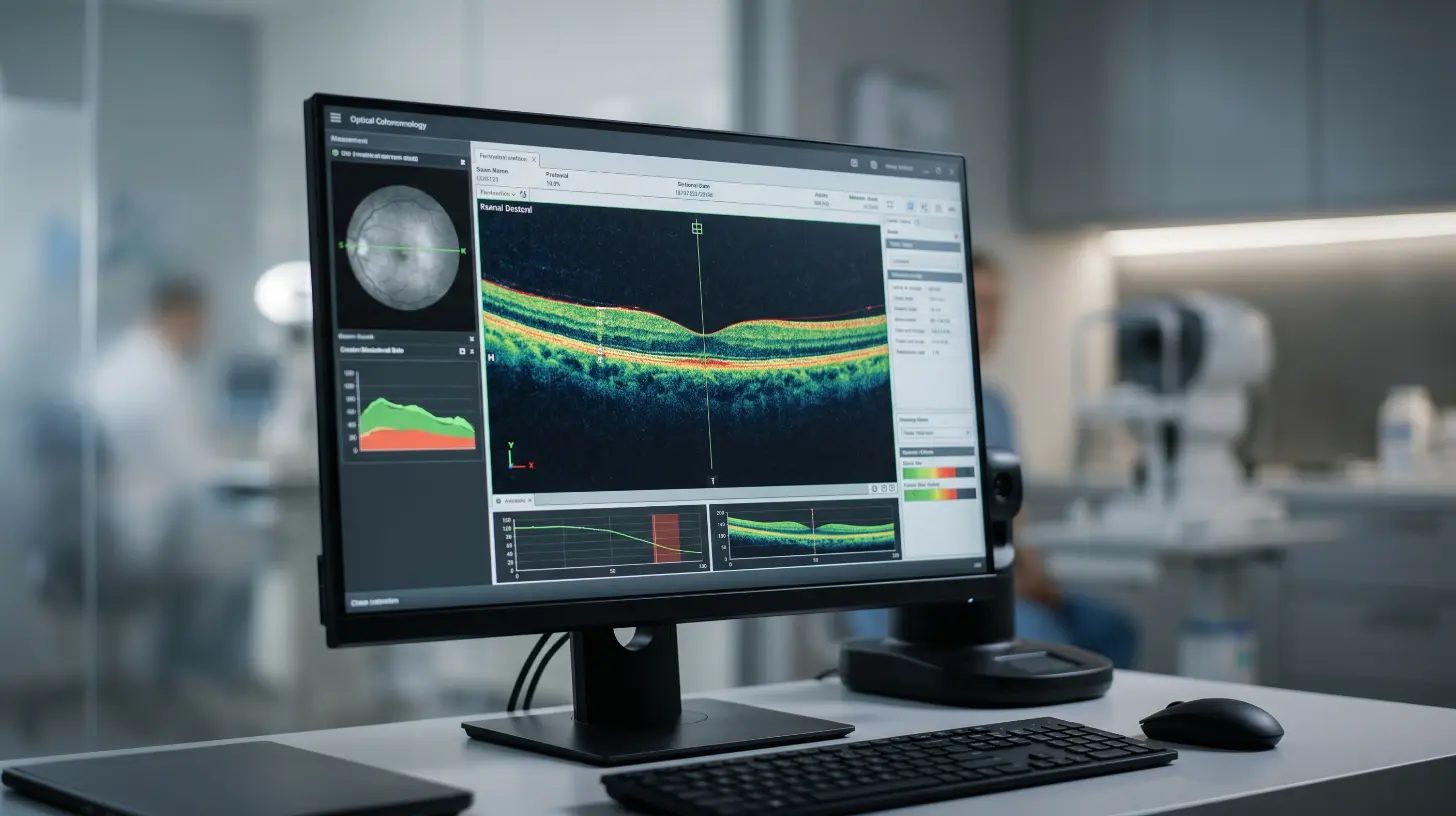
El diagnóstico requiere una evaluación especializada por un retinólogo con experiencia en enfermedades hereditarias. El Dr. Rodrigo Matsui, con formación en la Universidad de Pennsylvania y la Universidad de Heidelberg, utiliza un protocolo diagnóstico de precisión que incluye:
Existe un avance importante en el tratamiento de la retinosis pigmentaria, actualmente existen más de 300 ensayos clínicos que evalúan tratamientos para retinosis pigmentaria.
Las opciones disponibles incluyen manejo y protección visual:
La terapia génica representa el avance más significativo en el tratamiento de la retinosis pigmentaria. El tratamiento con voretigene neparvovec (Luxturna®), aprobado por la FDA, está indicado para pacientes con mutaciones bialélicas en el gen RPE65 y ha demostrado mejorar significativamente la función visual.
El Dr. Rodrigo Matsui es el primer médico en México con experiencia en la aplicación clínica de este tratamiento, formación adquirida en la Universidad de Pennsylvania bajo la mentoría del Dr. Samuel Jacobson, uno de los investigadores líderes en terapia génica retinal a nivel mundial.
Existen actualmente múltiples protocolos de investigación activos para diferentes mutaciones genéticas de retinosis pigmentaria. El Dr. Matsui, como investigador del SNI, mantiene vínculos activos con centros de investigación internacionales para ofrecer a sus pacientes acceso a los avances más recientes.
Esta es la pregunta que más frecuentemente hacen los pacientes y sus familias al llegar a consulta. La respuesta honesta es que no existe aún una cura definitiva para la mayoría de los casos, pero el panorama ha cambiado radicalmente en los últimos años.
La aprobación de la terapia génica para mutaciones RPE65, los ensayos activos para docenas de otras mutaciones, y el desarrollo de tecnologías como los chips retinales y la optogenética, representan un horizonte de posibilidades que hace apenas una década parecía lejano. El diagnóstico genético preciso es hoy más importante que nunca, porque determina si el paciente es candidato a alguno de estos tratamientos.
Como miembro del SNI, el Dr. Rodrigo Matsui basa su práctica en evidencia científica global. Fuentes de referencia:
Ubicación de la consulta: Verena Oftalmología - Acceso 1 Centro Médico ABC Campus Santa Fe - Carlos Graef Fernández 154, Torre 3, PB. Lomas de Santa Fe, CDMX. → Agenda tu cita con el Dr. Matsui